Help and Tutorial
ChemMine Tools is a free online service for analyzing and clustering small molecules by structural similarities, physicochemical properties or custom data types. This tutorial introduces the functionalities, data formats, methods and algorithms of this web service.
Browser Recommendation
This site is developed with recent (c. 2019) versions of major desktop web browsers (Chrome, Firefox, Safari) in mind. Some tools may be unavailable or may not function as expected with other browsers.
Free Support
Do you have any questions, bug reports, or suggestions on how we could make ChemMine tools more intuitive, powerful, or useful?
Index
- General Functionality
- Compound Import, Viewing and Format Interconversions
- Similarity Searching
- Similarity Comparisons
- Clustering and Data Mining
- Molecular Property Predictions
- Interacting with ChemMine Tools from R
- Theory: Descriptors, Similarity Measures and Clustering Schemes
- For Developers: Contribute a new tool
Users can import their custom compounds into the workbench of ChemMine Tools by drawing structures in the browser, copy and paste, from local files, or from a PubChem search. Subsequently, the imported compounds can be submitted from the workbench to the different analysis services.
- Workbench Overview
-
All compound structure data are organized in the compound workbench of ChemMine Tools. The workbench interface allows users to add, edit and remove compounds, and to view compound structure images in batches and to submit them to the other online services (see below).
- Compound Import
-
To import compounds into the workbench, users can choose from the following five options:
- SMILES Import
-
Users can import compound structures in the standard SMILES format. An overview of this format can be found here: Wikipedia: Simplified molecular-input line-entry system. A more comprehensive definition can be found here: Daylight Chemical Information Systems: SMILES - A Simplified Chemical Language.
- Structural Drawing
-
Users can draw a chemical structure directly in the browser.
- SDF Import
-
Users can import compound structures in the standard SDF format. An overview of this format can be found here: Wikipedia: Chemical table file. A more comprehensive definition can be found here: BIOVIA CTfile formats.
- PubChem CID Import
-
Users can import compound structures directly from PubChem by typing or pasting in a list of PubChem CIDs (one per line).
- PubChem Search
-
Alternatively, users can search the PubChem database with text or structure similarity searches and upload the identified compounds interactively to the workbench by clicking the "Add to Workbench" menus.
- Viewing of Compounds in Batches
-
Once compounds are imported into the workbench, the user can view them in list form, and optionally display structure images alongside each compound. Additional functionalities of the workspace interface are: add/edit/delete options and interactive structure similarity searches against PubChem.
- Format Interconversions
-
For reformatting purposes, all compounds imported into ChemMine Tools can be saved in SMILES or SDF formats. The "My Compounds" page contains buttons to download the entire workbench as single SMILES or SDF file. Clicking further to an individual compound page provides download links for single compounds.
- Search Overview
-
ChemMine Tools provides two powerful structural similarity search algorithms: EI and PubChem Fingerpint. EI Search is an ultra-fast search tool developed in house (Cao et. al. 2010). PubChem Fingerprint searching connects directly to the PubChem database, and therefore can return compounds only recently added to PubChem. Both tools accept five different types of input: SMILES strings, structural drawings, SDF, and similarity to existing compounds in the Workbench. PLEASE NOTE: The EI search tool has been temporarily disabled as we update it's database.
- Searching for Compounds Similar to Those in Workbench
-
After uploading compounds to the Workbench, browse to "My Compounds" and click "search similar compounds" next to your compound of choice (Fig. 2). This will send the SMILES string to the search tool where you can select either EI or PubChem Fingerprint search. Please note: the similarity search provided by PubChem uses substructure-based fingerprints, while the similarity tools in the clustering and similarity services of ChemMine Tools are based on atom pairs and maximum common substructures. For more details, please see the theory section at the end of this tutorial.
Each tool in ChemMine Tools is just a linux command line script, which can be written in any language. A .yaml file describes the input and output formats to the server. To contribute a new tool: (1) fork our repository on GitHub, (2) add in your tool to /tools/tool_scripts, and (3) perform a pull request to our "develop" branch. Please look at a few of the existing tools as examples for how to write the script and .yaml files.
Tools should be added here: https://github.com/TylerBackman/chemminetools/tree/develop/tools/tool_scripts
-
General Functionality
-
Compound Import, Viewing and Format Interconversions
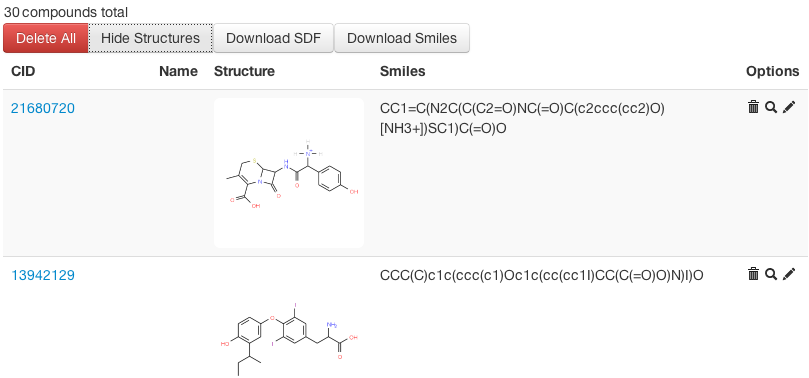
Fig 1: Compound View in Workbench -
Similarity Searching
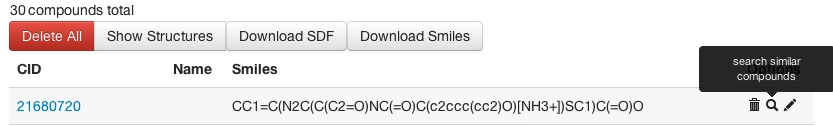
Fig 2: Search PubChem for Similar Compounds -
Similarity Comparisons
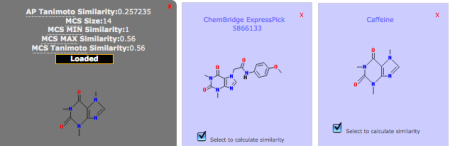
Fig 3: Calculation of Compound SimilaritiesSimilarity scores between compound pairs can be computed with the Similarity Workbench. The interface calculates atom pair and maximum common substructure (MCS) similarities with the Tanimoto coefficient as the similarity measure (Chen & Reynolds, 2002; Cao et al. 2008). The MCS tool identifies the largest substructure two compounds have in common (Fig. 3). Please note that the similarity measures provided by the Similarity Toolbox (MCS and AP) differ from PubChem fingerprint similarity, as used in the Search Toolbox. For more details, please see the theory section at the end of this tutorial.
-
Clustering and Data Mining
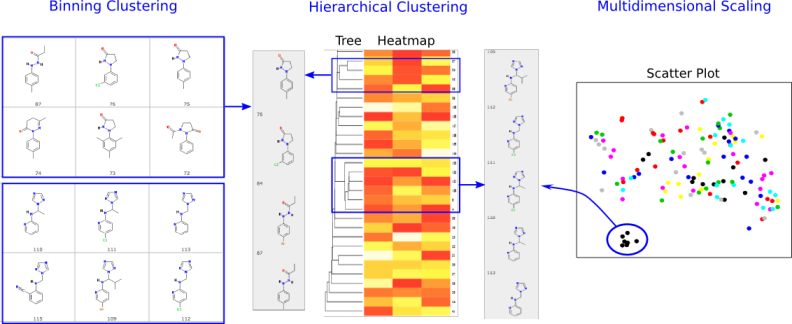
Fig 4:Binning, Hierarchical and Multidimensional Scaling ClusteringClustering of compounds by structural and physicochemical similarities is a powerful approach for correlating structural features of compounds with their activities. ChemMine Tools provides facilities for binning clustering, hierarchical clustering and multidimensional scaling (MDS). The required distance matrices for hierarchical and MDS clustering are calculated by all-against-all comparisons of compounds using atom pair similarity measures (see above) and transforming the generated similarity scores into distance values. The resulting trees and scatter plots are presented as interactive plots generated with the CanvasXpress javascript library. Trees and scatter plots can be zoomed and panned, and clicking a compound CID will pop-up a box with the compounds structure and a link to the compounds details page. The tree viewing tool can also plot heatmaps of generated physicochemical properties or data uploaded with the Upload Numeric Data link. This is useful for showing custom data like bioactivity information from HT screens in form of heatmaps next to the hierarchical clustering results.
-
Molecular Property Predictions
Fig 5: Molecular Property DescriptorsMolecular descriptors provide quantitative information about chemical properties of compounds. They can be very useful for prioritizing lead compounds, property clustering and QSAR analyses. Currently two property computing tools are provided- JOELib and Open Babel. The Open Babel tool computes the descriptors currently supported by the Open Babel software library. The JOELib tool computes 38 descriptors for each compound, which are described in detail here: JOELib site. After calculating molecular descriptors these data can be selected as input for various clustering jobs. To cluster based on properties, or plot a heatmap of your properties alongside a structural clustering tree: start a clustering job, and select your completed properties job from the drop-down menu.
-
Interacting with ChemMine Tools from R
Users can launch tools and obtain results programmatically using the R programming language. Instructions are provided in the ChemmineR library vignette.
-
Theory: Descriptors, Similarity Measures and Clustering Schemes
- Introduction
-
This section provides a brief overview of the cheminformatics and clustering algorithms used by ChemMine Tools. At the beginning of each subsection the services are listed in brackets [] where the corresponding methods and algorithms are used.
- Structure Similarity Comparisons and Searching of Small Molecules
-
To compare, cluster and search small molecules with respect to their structural similarities, a common approach is to enumerate their structural features, which are often referred to as structural descriptors. The numbers of common and unique features are then used to calculate a similarity measure among two compounds. The descriptor types and similarity coefficients used by ChemMine Tools are described below.
- (1) Structural Descriptors
- (a) Atom Pairs
- [ Similarity Comparison Clustering ]
-
Atom pairs are a structural descriptor type that is defined by the shortest paths among the non-hydrogen atoms in a molecule. Each path is described by the types of atoms in a pair, the length of their shortest bond path, the number of their pi electrons and the non-hydrogen atoms bonded to them. The number of atom pairs describing a molecule grows with its number of atoms. To use atom pairs for similarity comparisons, one can simply enumerate their common and unique atom pairs, and then use these numbers to compute a similarity coefficient (see below).
- (b) PubChem Fingerprints
- [ Similarity Search ]
-
The fingerprints provided by PubChem are a binary representation of the presence and absence of a library of 881 substructure features (see here for details). In this system every molecular structure is described by 881 bits where 1 indicates the presence and 0 the absence of a feature. Compared to atom pairs, the PubChem fingerprints are a knowledge-based system that stores less information than the much more complex and unbiased atom pair concept. For database searching fingerprints are often much more time and memory efficient, but they are less sensitive than atom pair descriptors (see Chen & Reynolds, 2002; Cao et al. 2008).
- (c) Maximum Common Substructure
- [ Similarity Comparison ]
-
The maximum common substructure (MCS) problem is a graph-based similarity concept that is defined as the largest substructure (sub-graph) shared among two compounds. It is a pair-wise concept that is not directly related to the above structural descriptors, but its results (e.g. size of MCS relative to source structures) can be used for the computation of the same similarity coefficients (see below). Compared to descriptor-based similarity concepts, the MCS method provides the most accurate and sensitive similarity measure, especially for compounds with large size differences.
- (2) Similarity Coefficients
- (a) Tanimoto coefficient
- [ Similarity Comparison Similarity Search Clustering ]
-
The Tanimoto coefficient is defined as c/(a+b+c), which is the proportion of the features shared among two compounds divided by their union. The variable c is the number of features (or on-bits in binary fingerprint) common in both compounds, while a and b are the number of features that are unique in one or the other compound, respectively. The Tanimoto coefficient has a range from 0 to 1 with higher values indicating greater similarity than lower ones. It is important to emphasize that a Tanimoto coefficient of 1 does not necessarily mean that two compounds are identical. It only means that they have identical structural descriptors or identical on-bits in a binary fingerprint. To determine identity among two compounds, InChI strings usually provide a much more reliable solution. This latter feature will become available in ChemMine Tools soon.
- (b) Tversky Index
- [ Similarity Comparison ]
-
The Tversky index is defined as c/(α*a + β*b + c). It extends the Tanimoto index by two weighting variables α and β. If α and β are set to 1 then the index returns the same result as the Tanimoto coefficient.
- (c) Dice Index
- [ Similarity Comparison ]
-
Setting α and β in the Tversky index to 0.5 returns the Dice index.
- Similarity Measures for Property and Activity Profiles
- [ Clustering ]
-
Sets of numeric property values of compounds, such as physiochemical properties or bioactivity values, can also be used to compute a similarity measure among compounds. For instance, ChemMine Tools uses the physiochemical descriptors of compounds - or any numeric custom data set - for the computation of Pearson correlation coefficients as similarity measure for the calculation of an item-to-item similarity matrix that can be converted into a distance matrix for downstream clustering.
Clustering based on property values is performed follows:
- Scaling and centering the property table row-wise by subtracting from each value the row mean and then dividing by the standard deviation of each row.
- Calculation of an all-against-all Pearson correlation matrix. This is transformed into a distance matrix by subtracting each value from 1.
- Hierarchical clustering of this distance matrix.
- Clustering Methods and Schemes
- (a) Hierarchical Clustering
- [ Clustering ]
-
This services uses the hclust function implemented in R to perform hierarchical clustering. It requires as input a distance matrix of all-against-all compound distances that is generated by subtracting the similarity measure (e.g. Tanimoto coefficient Tc) from one (1 - Tc). The resulting distance matrix is then passed on to the actual clustering program that hierarchically joins the most to least similar items in an agglomerative manner using as cluster joining rule either single, average or complete linkage. The latter parameters are definable by the user.
- (b) Multidimensional Scaling
- [ Clustering ]
-
Similar to hierarchical clustering, multidimensional scaling (MDS) starts with a matrix of item-item distances and then assign coordinates for each item in a low-dimensional space to represent the distances graphically in a scatter plot. The cmdscale function implemented in R is used for this service.
- (c) Binning Clustering
- [ Clustering ]
-
Binning clustering assigns compounds to similarity groups based on a user-definable similarity cutoff. For instance, if a Tanimoto coefficient of 0.6 is chosen then compounds will be joined into groups that share a similarity of this value or greater using a single linkage rule for cluster joining. This method is based on an internally developed C++ implementation that is very memory efficient since it does not require a distance matrix as input. It calculates the required compound-to-compound distance information on the fly.
-
For Developers: Contribute a new tool